![]()
![]()
![]()
病院レター第114号 2025年1月1日

脳神経内科部長 新畑豊
「高齢になり動きが悪くなり転ぶことが増えた、パーキンソン病(PD)ではないか」とご紹介いただく機会が増えてきています。多くの医療者の間で動きが悪くなる病気として「パーキンソン病」という病名が浸透してきたからだと思われます。PDは大まかにはレビー小体の出現を病理学的特徴とする単一疾患ですが、PDに類似した動きの鈍さを持つ病態を総称的にパーキンソン症候群(パーキンソニズム)と呼びます。これらには一体どのような疾患や特徴があるのかを解説します。
PDは中脳黒質のドパミンを作る細胞が変性脱落することにより黒質-線条体神経の障害がおこり運動障害などが発症する疾患です。線条体でのドパミンが20%程度以下になると運動障害が引き起こされると考えられています。ドパミンを受け取る線条体の側の節後神経は脱落していないため、ドパミン補充療法(L-DOPA)が有効です。PDの治療薬としては、L-DOPA以外にも多種類の薬剤が開発され臨床に用いられており、治療により長期にわたる運動改善が期待できます。当院の外来に通院されている独歩が可能なレベルのPD患者さん約260人の平均の罹病期間(±標準偏差)は8.2(±6.5)年で15年以上の経過の方が16%程度おられます。
病理学的には変性した神経細胞の中にレビー小体と呼ばれるα-シヌクレインからなる特殊な構造物(封入体)がみられるという特徴があります。認知症を引き起こすレビー小体型認知症は、PDと同一のスペクトラム上にある疾患で神経変性の広がりなどが異なります。PDの運動症状は振戦、無動、固縮(筋強剛)、姿勢反射障害が4大症状です。PDに見られる振戦は「安静時振戦」が特徴で、何もせずじっとしているときにみられ、運動を開始することで一旦停止する特徴があります。振戦は特に高齢発症のPDでは見られない場合もあります。固縮は「歯車様固縮」といわれる、診察時に関節を動かすとカクカクとした強弱の変動をもつ抵抗があるのが特徴的です。PDの体の動きは全体に小さくなり、前屈姿勢になりがちで歩行時には歩幅は前後に狭い(小歩)とともに、左右の開脚も狭く、腕の振りも小さくなり、全体にコンパクトとなるといった特徴があります(ここでは、「PDらしさ」と表現します)。症状には通常左右の差があり、黒質―線条体神経の変性の左右差に対応します。PDの患者さんが発症当初から転倒を起こすことはまれで、早期の段階から頻回に転倒を起こす場合には、他の疾患を念頭に置く必要があります
一見PDに類似する動作緩慢、歩幅が狭くなる(小歩症)、すり足、筋強剛(固縮)などの症状がみられるものを総じてパーキンソン症候群(パーキンソニズム)と呼ぶことがあります。パーキンソン症候群の主な原因には、薬剤性、中毒性、脳血管性パーキンソニズム、正常圧水頭症、PD以外の脳変性疾患(進行性核上性麻痺、大脳皮質基底核変性症、多系統萎縮症など)があります。これらの多くは、PD治療薬が無効か、効果があったとしても限定的です。逆に、抗PD薬であるL-DOPAの治療効果がはっきりあった場合には、本来のPDである可能性が高く、治療をきちんとすることで予後が改善できる可能性があるため、効果を見極める必要があります。
ドパミン受容体をブロックする作用を持つ薬剤の投与によりパーキンソニズムが引き起こされることがあります。PDとは異なり、左右均等に症状が出ることが多く、振戦を伴う場合は、安静時振戦ではなく、動作時の振戦であることが多くみられます。古くからある抗精神病薬であるハロペリドール、クロルプロマジンなどは原因となる薬剤としてよく知られていますが、第二世代の抗精神病薬であるリスペリドンなどでもそのリスクはあります。また、胃腸薬としても用いられるスルピリドは汎用性の高さからか薬剤性パーキンソニズムとして来られる患者さんの原因薬剤としては高いものと考えられています。中には、PDの準備状態にある(おそらく黒質-線条体神経の脱落があるが、自覚する運動障害がなかった状態)の患者さんにこれらの薬剤が投与され、PDの症状が誘発されたと考えられる例もあり、慎重な鑑別が必要となる場合があります。この場合、ドパミントランスポーターSPECT検査(123I-Ioflupane;ダットスキャンⓇ)で黒質線条体神経の変性があるかどうかを見ることが、診断の大きな手掛かりになります。
古くから「脳血管性パーキンソニズム」という言葉はありますが、その実態はあまりはっきりしていません。脳のCTやMRI検査で大脳基底核部分の多発ラクナ梗塞や、特に前頭葉の広汎な白質障害をきたした例(Binswanger型白質脳症)がみられる例に動作緩慢、小歩などの症状がみられることがあり、これがパーキンソニズムととらえられたものが多いと思われます。実際には、前頭葉障害による失行(うまく行動ができない)や両側の不全麻痺、動作の緩慢さといった症状が複合した病態と考えられます。固縮はあっても鉛管様といわれる一様の抵抗を感じるものが多く、歩行は前後に狭いすり足ですが、左右は開脚(wide-based)、上肢の振りの制限も目立たず、前屈姿勢もとることは少ないため、PDのコンパクトな運動とは様相が異なります。脳のCTやMRIで多数の梗塞巣がみられても、「PDらしさ」を持つ症状がある場合にはPDである可能性があるため、治療効果を見極める必要があります。
髄液の産生と吸収のアンバランスにより、脳室拡大をはじめとするDisproportionately enlarged subarachnoid space hydrocephalus (DESH)と呼ばれる特徴的なCTやMRI画像所見がみられ、認知機能低下、尿失禁などがみられやすい病態です。多くの場合、髄液を脳以外へ逃がすシャント手術が有効です。歩行障害はwide-basedな小股歩行となりやすく、やはり「PDらしさと」は異なるものが多いようです。逆にDESHの所見があり「PDらしさ」があるものは合併を疑う必要があります。
ドパミン神経の節後部の神経脱落が起こるため、基本的にドパミン補充療法が無効であり、PDに比べ運動障害の進行が速いものがほとんどです。ドパミン神経節前部を評価するダットスキャンⓇの検査ではいずれもPD類似の異常を示すため、心臓交感神経の節後線維の異常を表すMIBG心筋シンチグラムがPDとの鑑別(PDでのみ異常がみられやすい)に有効です。
古典的に知られる症状のPSPは「リチャードソン症候群」と呼ばれ、発症の初期より姿勢反射障害が強く、繰り返し転びやすいことが特徴です。歩行は開脚気味ですくみ足や加速歩行といった現象もみられすいです。左右差が乏しく手足よりも頸部や体幹の固縮がみられます。進行すると頸部が後屈気味となり、眼球運動の上下への運動制限がみられやすいため、足元を見にくく、呂律や嚥下の障害も出やすくなります。前頭葉の機能障害による認知機能の障害を伴いやすいため、抑制が効きにくい突発的な行動があるため、運動機障害だけではない側面から転倒に結び付きやすいこともあります。病理学的なPSPには臨床表現型としては亜型があり、初期にはL-DOPAに反応する「PDらしさ」を併せ持つもの、すくみ足だけを症状とするもの、小脳失調を中核症状とするもの、前頭側頭型認知症類似の症状を呈するものなどがある事が知られています*1。基本的には長期のL-DOPA治療効果は期待できず、寝たきりになるまでの平均予後は5~7年とされます。病理学的には異常リン酸化タウ蛋白が蓄積する4リピートタウオパチーです。
臨床的な典型例は「皮質基底核症候群(CBS)」と呼ばれる症状を呈し、左右非対称の固縮や無動、ミオクローヌス(震えの一種)、ジストニア(姿勢異常)などとともに大脳皮質の障害の症状として、種々の失行、強制把握、他人の手徴候(自分の意思と関係なく腕や手が勝手に動く)といった症状がみられ、PDの鑑別が必要な疾患の一つに数えられます。病理学的にはPSPと同様の4リピートタウオパチーに分類されます。病理学的な広がりの違いにより、失語が主体の型、前頭側頭型認知症類似型、PSP類似の型などがしられ*2、これらの亜型を臨床診断することは難しい場合もしばしばです。
病理学的には小脳、橋、線条体、黒質、脳幹や脊髄の自律神経核の神経細胞の変性がみられる疾患です。グリア細胞にα-シヌクレインからなる封入体(GCI)が出現する特徴があります。初期に主体となる臨床症状により、パーキンソニズムを主体とするMSA-P(線条体黒質変性症)、小脳失調とを主体とするMSA-C(オリーブ橋小脳変性症)、起立性低血圧や排尿障害といった自律神経障害を中心とするシャイ・ドレーガー症候群に分類されます。MRIで線条体や橋、小脳の萎縮がみられるとPDとの鑑別につながります*3。
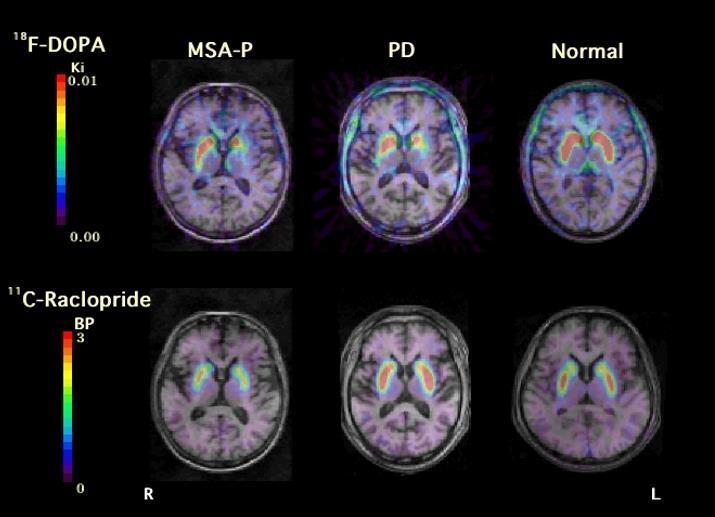
図:多系統萎縮症(MSA-P)、パーキンソン病(PD)、正常者(Normal)のドパミン神経系のPET評価 (*3より図を引用)
18F-DOPAはドパミン神経に取り込まれ、線条体でのドパミン神経(節前部)終末の働きを表し、11C-Racroprideは線条体神経(節後部)のドパミンD2受容体密度を表す。図は同一被検者の脳MRIとの重ね合わせ画像。PD、MSAは類似の被殻の後方に優位なドパミン神経の障害があるが、MSAでは節後神経の脱落がある事を示している。PD例は未治療例であるため、節後部D2受容体の正常よりの活性亢進が示されている。(いずれも当センターでの自験例、PET検査はいずれも保険適用外であり研究として実施されたもの)
現在18F-DOPA PETに代わり、臨床的にはほぼ等価のドパミン節前神経密度を見るためにドパミントランスポーターを見るSPECT検査(123I-Ioflupane SPECT:ダットスキャンⓇ)が保険収載され実施可能であるが、空間分解能がPETの半分程度であるため、線条体内部構造を分離した評価まではむつかしい。
パーキンソン症候群と一口に言っても、その症状は多彩で、背景にも多数の疾患の可能性があります。「PDらしさ」を見抜いて、有効な治療に結び付けることが必要です。PD以外のものは、現状で有効な薬剤治療が乏しいものが多いのですが、状態に応じたQOLの維持に有効な方法を相談・提案していくことが重要かと考えています。
- Williams DR, Lancet Neurol. 2009;8(3):270-
- Armstrong MJ, Neurology. 2013;80(5):496-
- 新畑豊 CLINICALNEUROCIENSE 2006.24(9):1017-
長寿医療研究センター病院レター第114号をお届けいたします。
パーキンソン病は、神経変性疾患の一つであり、神経変性疾患はある特定の神経細胞群が、徐々に障害を受けて脱落してしまう疾患です。パーキンソン病の場合は、α-シヌクレインの凝集が長い年月をかけて起こり、発症するとされていますが、本稿でも解説されているようにパーキンソン病と類似の症候を示すPSPやCBDは異常リン酸化タウの蓄積が起こるとされています。タウ蛋白は細胞骨格の一つである微小管に結合する蛋白であり、微小管の重合促進と安定化に働き、正常では神経細胞の軸索に局在します。タウ蛋白の異常蓄積が始めて発見されたのはアルツハイマー病であり、異常なタウ蛋白が蓄積する疾患をタウオパチーと呼びます。タウ蛋白には6つのアイソフォームがあり、一つの遺伝子から選択的スプライシングによって作られます。6つのアイソフォームは微小管結合部位の繰り返し配列構造が3つの3リピートタウと4つの4リピートタウに大別されます。PSP、CBDおよび嗜銀性顆粒認知症では4リピートタウが蓄積し、また前頭側頭変性症の一部(ピック病)では3リピートタウが蓄積するとされています。さらにこれら3リピートタウと4つの4リピートタウの6つのアイソフォームすべてが蓄積するのがアルツハイマー病です。
現在アルツハイマー病の診断および進行度、さらには治療効果の評価のためにPETを使って脳内のタウ蛋白の蓄積状況を明らかにする研究が盛んに行われており、当センターでもタウPET薬剤を使って、様々なタウオパチーでタウ蛋白に薬剤が結合するかどうかを確認する研究を行っています。将来的には、タウPET薬剤が臨床医療で大きな役割を果たす時代が確実にやってきます。

今後も引き続き、パーキンソン病および類似疾患、さらには認知症の研究および臨床の最新情報を発信して行きたいと考えておりますので、何卒よろしくお願いいたします。
病院長 近藤和泉



![]()
© National Center for Geriatrics and Gerontology

